Glutaric Aciduria type I (GA1) is a rare organic acid disorder with estimated overall prevalence of 1 in 100,000 newborns.
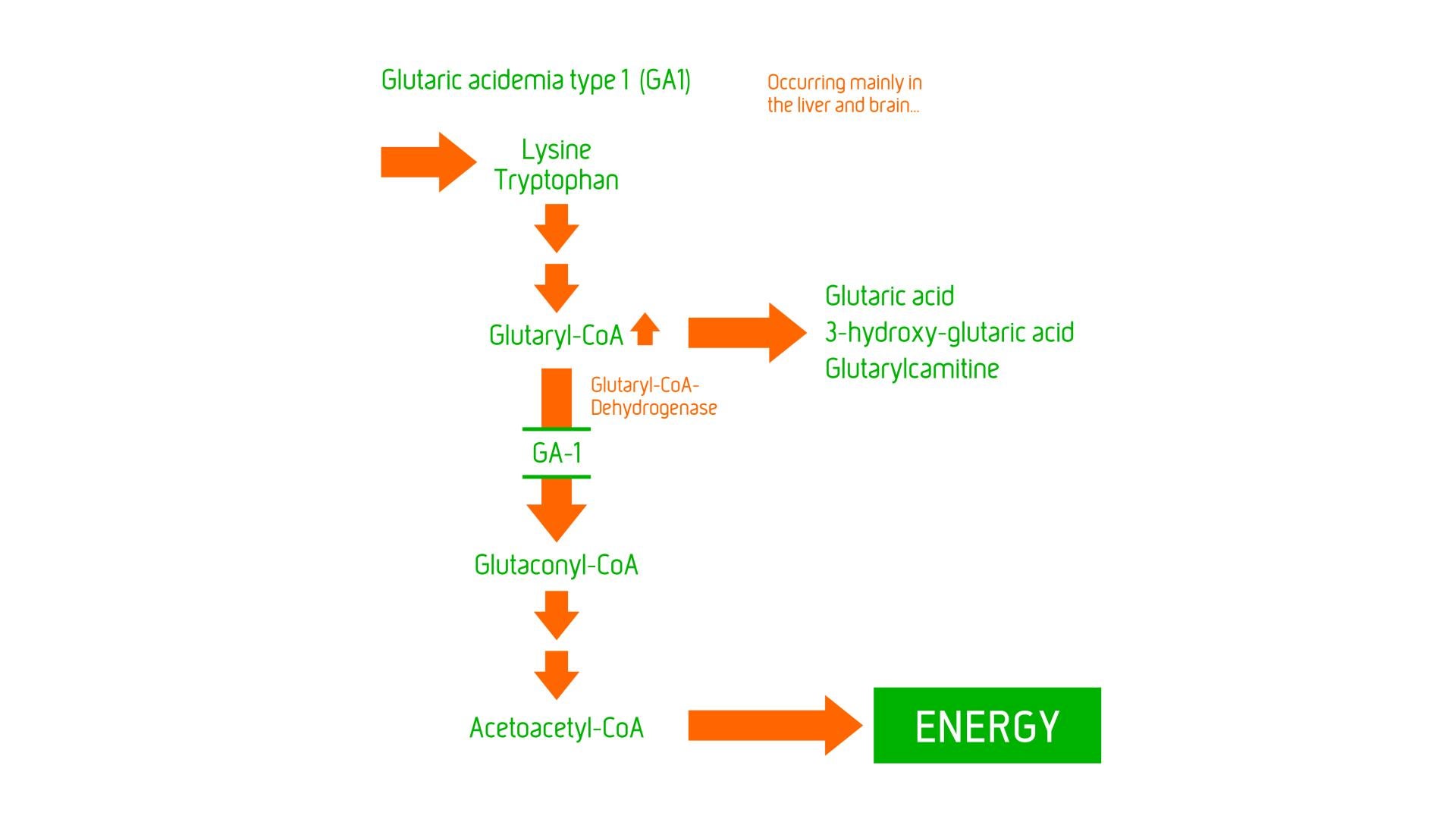
GA1 is considered to be a neurometabolic disorder, caused by glutaryl-CoA dehydrogenase deficiency resulting in problems with the ability to break down amino acids – lysine, hydroxylysine and tryptophan, leading to excessive levels of their intermediate metabolic products – (neurotoxic) glutaric acid and 3-hydroxyglutaric acid and (non-toxic) glutarylcarnitine in body fluids. The toxic chemicals can cause brain injury, particularly in the basal ganglia (an area of the brain that regulates motor movement) resulting in a severe movement disorder – dystonia.
GA1 is now regarded as a treatable metabolic disorder and is included in the disease panel of expanded newborn screening in some countries.
With treatment, normal outcome is possible. Without treatment, outcomes vary but many will have an acute encephalitis-like crisis that may result in developmental delay, neurologic deterioration and dystonia. Otherwise, babies born apparently healthy could then develop macrocephaly, sometimes with hypotonic, if untreated.
Acute encephalopathic crises usually occur between 6 and 18 months of age and typically are triggered by febrile illness, immunization, surgery, and fasting. After 5-6 years of age, the risk of acute decompensation is low.
In the majority of patients diagnosed in the neonatal period, brain damage can be prevented by metabolic treatment. This includes dietary treatment in combination with L-carnitine supplementation and emergency treatment during acute episodes of intercurrent illness.
Dietary treatment aims to reduce the intake of lysine and tryptophan, while maintaining sufficient intake of essential nutrients, minerals, vitamins and trace elements.
To avoid malnutrition the diet is often combined with the use of lysine-free, tryptophan-reduced amino acids supplements such as comida-GAc A formula.

